PCR技术起源
聚合酶链反应( PCR ) 是一种广泛用于快速制造特定DNA样本数百万到数十亿份(完整拷贝或部分拷贝)的方法,使科学家能够获取非常小的 DNA 样本并对其(或部分 DNA 进行扩增)。它)足够大的数量来详细研究。PCR是由美国在1983年发明的生化学家 凯利·穆利斯在鲸鱼座公司。它是基因检测和研究中使用的许多程序的基础,包括分析古代 DNA 样本和识别传染源。使用 PCR,复制极少量的DNA 序列在一系列温度变化循环中呈指数放大。PCR 现在是医学实验室研究中常用且通常不可或缺的技术,其应用范围广泛,包括生物医学研究和刑事取证。
大多数 PCR 方法依赖于热循环。热循环将反应物暴露于重复的加热和冷却循环中,以允许不同的温度依赖性反应——特别是DNA 熔解和酶驱动的DNA 复制。PCR 使用两种主要试剂——引物(它们是短的单链 DNA 片段,称为寡核苷酸,是目标 DNA 区域的互补序列)和DNA 聚合酶。在 PCR 的第一步中,DNA 双螺旋的两条链在称为核酸变性的过程中在高温下物理分离. 第二步,降低温度,引物与 DNA 的互补序列结合。然后,两条 DNA 链成为DNA 聚合酶的模板,以酶促方式从游离核苷酸(DNA 的构建块)组装一条新的 DNA 链。随着 PCR 的进行,生成的 DNA 本身用作复制模板,启动链式反应,其中原始 DNA 模板呈指数放大。
几乎所有 PCR 应用都使用热稳定 DNA 聚合酶,例如Taq聚合酶,这种酶最初是从嗜热细菌Thermus Aquaticus 中分离出来的。如果使用的聚合酶是热敏感的,它会在变性步骤的高温下变性。在使用Taq聚合酶之前,每个循环都必须手动添加 DNA 聚合酶,这是一个繁琐且成本高昂的过程。
该技术的应用包括用于测序的DNA克隆、基因克隆和操作、基因诱变;基于 DNA 的系统发育的构建,或基因的功能分析;诊断和监测的遗传性疾病; 古代DNA的扩增;用于DNA分析的基因指纹分析(例如,在法医学和亲子鉴定中);和检测的病原体中核酸测试用于诊断感染性疾病。
PCR发展历史
作为聚合酶链反应关键组成部分的耐热酶是在 1960 年代发现的,它是生活在黄石蘑菇泉过热水中的微生物生命形式的产物。
1971 年,Kjell Kleppe和H. Gobind Khorana实验室的同事在《分子生物学杂志》上发表的一篇论文首先描述了一种使用酶促测定法在体外复制带有引物的短 DNA 模板的方法。然而,这种基本 PCR 原理的早期表现在当时并未受到太多关注,1983 年聚合酶链反应的发明通常归功于Kary Mullis。
当穆利斯于 1983 年开发 PCR 时,他正在加利福尼亚州埃默里维尔为第一批生物技术公司之一的Cetus Corporation 工作,负责合成 DNA 短链。穆利斯写道,他在驾驶汽车沿着太平洋海岸公路巡航时构思了 PCR 的想法。当他意识到自己发明了一种通过 DNA 聚合酶驱动的重复复制循环来放大任何 DNA 区域的方法时,他正在考虑一种分析 DNA 变化(突变)的新方法。在科学美国人,穆利斯总结了这个过程:“从遗传物质 DNA 的单个分子开始,PCR 可以在一个下午产生 1000 亿个类似的分子。反应很容易执行。它只需要一个试管,几个简单的试剂,以及热源。” DNA指纹识别于1988年首次用于亲子鉴定。
Mullis 将LSD 的使用归功于他开发 PCR 的一个组成部分:“如果我没有服用 LSD,我会发明 PCR 吗?我严重怀疑它。我可以坐在 DNA 分子上看着聚合物经过。我学到了部分原因是迷幻药物。”
穆利斯和迈克尔·史密斯教授开发了其他重要的 DNA 操作方法,于 1993 年共同获得了诺贝尔化学奖,距离穆利斯和他在 Cetus 的同事们第一次将他的提议付诸实践七年。 Mullis 1985 年与 RK Saiki 和 HA Erlich 发表的论文“β-珠蛋白基因组序列的酶促扩增和用于诊断镰状细胞贫血症的限制性位点分析”——聚合酶链反应发明 (PCR)——获得化学引文奖2017年美国化学会化学史分会突破奖。
PCR 方法的核心是使用合适的DNA 聚合酶,该酶能够承受在每个复制循环后分离DNA 双螺旋中的两条 DNA 链所需的 >90 °C (194 °F) 的高温。最初用于PCR 前体外实验的 DNA 聚合酶无法承受这些高温。因此,早期的 DNA 复制程序非常低效且耗时,并且在整个过程中需要大量的 DNA 聚合酶和连续处理。
在1976年发现的Taq聚合酶-a DNA聚合酶从纯化的嗜热菌,栖热菌属aquaticus,这自然生活在热的(50至80℃(122〜176°F))的环境中[13]如温泉-铺平了显着改进 PCR 方法的方法。从T.aquaticus 中分离的 DNA 聚合酶在高温下是稳定的,即使在 DNA 变性后仍保持活性,因此无需在每个循环后添加新的 DNA 聚合酶。这使得基于自动热循环仪的 DNA 扩增过程成为可能。
专利纠纷
PCR 技术由Kary Mullis申请专利,并转让给Cetus Corporation,Mullis 于 1983 年发明该技术时曾在该公司工作。Taq聚合酶也受专利保护。已经有几起与该技术相关的备受瞩目的诉讼,其中包括杜邦公司提起的一起败诉。瑞士制药公司Hoffmann-La Roche于 1992 年购买了这些专利的权利,目前[什么时候?]保存那些仍然受到保护的。
罗氏和Promega之间在世界各地的几个司法管辖区仍在进行有关Taq聚合酶的专利之战。法律争论已经超出了原 PCR 和Taq聚合酶专利的有效期,这些专利于 2005 年 3 月 28 日到期。
PCR技术原理图
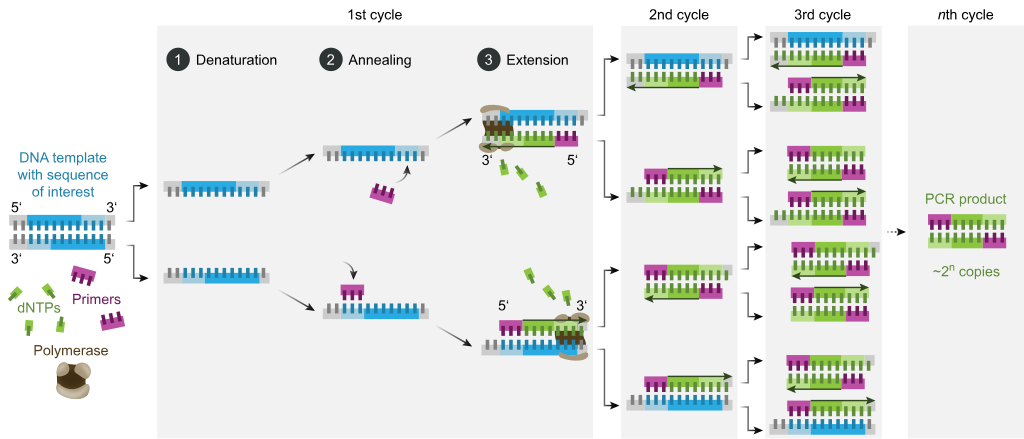
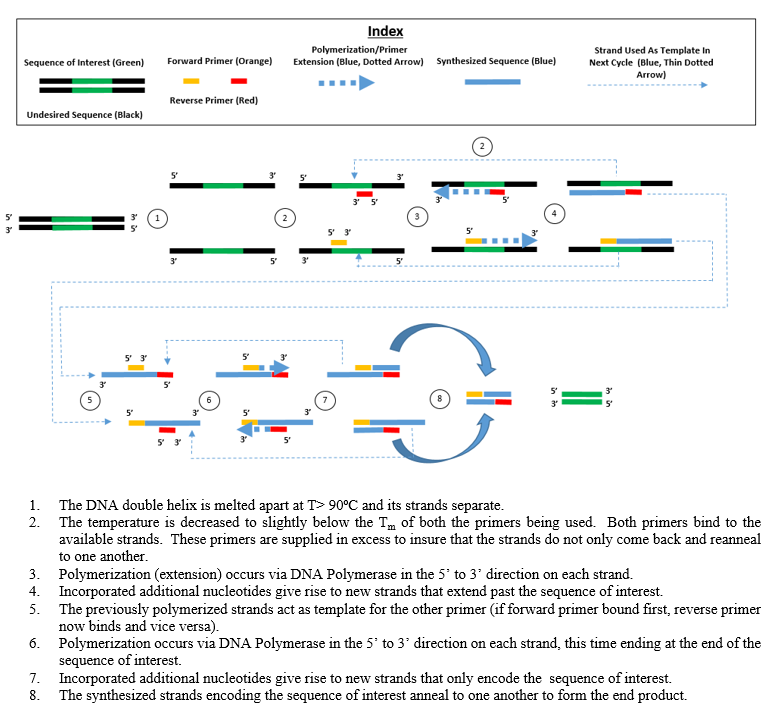
通常,PCR 由一系列 20-40 次重复的温度变化组成,称为热循环,每个循环通常由两个或三个离散的温度步骤组成(见下图)。循环之前通常是在非常高的温度(> 90 °C (194 °F))下进行单个温度步骤,然后在最后进行一次保温以延长最终产品或进行短暂储存。在每个循环中使用的温度和它们应用的时间长度取决于各种参数,包括用于 DNA 合成的酶、反应中二价离子和 dNTP 的浓度以及熔解温度( T m )引物。大多数 PCR 方法共有的各个步骤如下:
初始化:此步骤仅适用于需要通过热启动 PCR进行热激活的 DNA 聚合酶。它包括将反应室加热到 94–96 °C (201–205 °F) 或 98 °C (208 °F)(如果使用极其耐热的聚合酶),然后保持 1– 10分钟。
变性:此步骤是第一个常规循环事件,包括将反应室加热至 94–98 °C (201–208 °F) 20–30 秒。这通过破坏互补碱基之间的氢键导致双链 DNA 模板的DNA 解链或变性,产生两个单链 DNA 分子。
退火:下一步,将反应温度降低至 50–65 °C (122–149 °F) 20–40 秒,使引物与每个单链 DNA 模板退火。反应混合物中通常包含两种不同的引物:一种用于包含目标区域的两条单链互补序列中的每一条。引物本身是单链序列,但比目标区域的长度短得多,仅与每条链 3' 端的非常短的序列互补。
为退火步骤确定合适的温度至关重要,因为效率和特异性受退火温度的强烈影响。该温度必须足够低以允许引物与链杂交,但又足够高以使杂交具有特异性,即引物应仅与链的完美互补部分结合,而不能与其他任何地方结合。如果温度太低,引物可能无法完美结合。如果它太高,引物可能根本不会结合。典型的退火温度比T m低约 3–5 °C使用的引物。只有当引物序列与模板序列非常匹配时,互补碱基之间才会形成稳定的氢键。在此步骤中,聚合酶与引物-模板杂交体结合并开始形成 DNA。
延伸/延伸:这一步的温度取决于所用的 DNA 聚合酶;Taq聚合酶的热稳定 DNA 聚合酶的最佳活性温度约为 75–80 °C (167–176 °F),尽管此温度通常使用 72 °C (162 °F)酶。在此步骤中,DNA 聚合酶通过从反应混合物中加入与模板在5' 到 3' 方向互补的游离 dNTP ,缩合5'-磷酸基团,从而合成与 DNA 模板链互补的新 DNA 链具有 3'-羟基的 dNTP在新生(伸长)DNA 链的末端。延伸所需的精确时间取决于所用的 DNA 聚合酶和要扩增的 DNA 目标区域的长度。根据经验,在最佳温度下,大多数 DNA 聚合酶每分钟聚合 1000 个碱基。在最佳条件下(即,如果由于底物或试剂的限制而没有限制),在每个延伸/延伸步骤中,DNA 目标序列的数量加倍。随着每个连续循环,原始模板链加上所有新生成的链成为下一轮延伸的模板链,导致特定 DNA 目标区域的指数(几何)扩增。
变性、退火和伸长的过程构成一个循环。需要多次循环才能将 DNA 目标扩增到数百万个拷贝。用于计算给定循环数后形成的 DNA 拷贝数的公式是 2 n,其中n是循环数。因此,设置 30 个循环的反应产生 2 30或 1,073,741,824 个原始双链 DNA 目标区域的拷贝。
最终延伸:这一步是可选的,但在 70–74 °C (158–165 °F)(PCR 中使用的大多数聚合酶的最佳活性所需的温度范围)的温度下进行 5–15 分钟。最后一个 PCR 循环,以确保任何剩余的单链 DNA 都被完全拉长。
最后保持:最后一步将反应室无限期冷却至 4–15 °C (39–59 °F),并可用于 PCR 产物的短期储存。
为了检查 PCR 是否成功生成了预期的 DNA 目标区域(有时也称为扩增子或扩增子),可以采用琼脂糖凝胶电泳对 PCR 产物进行大小分离。PCR 产物的大小是通过与DNA分子量标准比较来确定的,DNA分子量标准是一种分子量标记,包含已知大小的 DNA 片段,与 PCR 产物一起在凝胶上运行。
-
与其他化学反应一样,PCR 的反应速率和效率受到限制因素的影响。因此,整个 PCR 过程可以根据反应进程进一步分为三个阶段:
- 指数扩增:在每个循环中,产物量加倍(假设反应效率为 100%)。30 个循环后,单个 DNA 拷贝可以增加到 1,000,000,000(十亿)个拷贝。从某种意义上说,DNA 离散链的复制是在受控条件下在管中进行的。该反应非常敏感:只有微量的 DNA 必须存在。
- 平稳阶段:随着 DNA 聚合酶失去活性以及试剂(如 dNTP 和引物)的消耗,反应会变慢,导致它们变得更加有限。
- 消耗: 由于试剂和酶的耗尽,没有更多的产品积累。
PCR优点与限制
优点
PCR具有许多优点。它非常易于理解和使用,并且可以快速产生结果。该技术高度敏感,有可能产生数百万到数十亿份用于测序、克隆和分析的特定产品。qRT-PCR 与 PCR 具有相同的优势,但具有合成产物定量的额外优势。因此,它可用于分析肿瘤、微生物或其他疾病状态中基因表达水平的变化。
PCR 是一种非常强大且实用的研究工具。许多疾病的未知病因的测序正在通过 PCR 计算出来。该技术可以帮助识别与已知病毒相关的先前未知病毒的序列,从而让我们更好地了解疾病本身。如果可以进一步简化程序并开发灵敏的非辐射检测系统,那么 PCR 将在未来几年内在临床实验室中占据重要地位。
限制
PCR 的一个主要限制是关于目标序列的先验信息是必要的,以便生成允许其选择性扩增的引物。这意味着,通常情况下,PCR 用户必须知道两个单链模板中每个目标区域上游的精确序列,以确保 DNA 聚合酶正确结合引物-模板杂交体和随后在 DNA 合成过程中生成整个目标区域。
与所有酶一样,DNA 聚合酶也容易出错,从而导致生成的 PCR 片段发生突变。
PCR 的另一个限制是,即使是最少量的污染 DNA 也可能被扩增,从而导致误导或模棱两可的结果。为了尽量减少污染的机会,研究人员应为试剂制备、PCR 和产品分析预留单独的房间。试剂应分装成一次性使用的等分试样。应经常使用带有一次性柱塞和超长移液器吸头的移液器。此外,建议确保实验室设置遵循单向工作流程。PCR 室和分析室中使用的任何材料或试剂都不得在未彻底净化的情况下带入 PCR 制备室。
含有腐殖酸的环境样品可能会抑制 PCR 扩增并导致结果不准确。